Research Topics
Dissecting the dynamics of protein interactions and complexes at their native loci with structural details
-
Mapping protein-protein interactomes across a range of species (S. cerevisiae, S. pombe, and humans)
By investigating these interactomes, we aim to elucidate conserved mechanisms and gain insights into species-specific protein interactions, ultimately advancing our understanding of evolutionary principles governing interactome networks, as well as various cellular processes and their regulation.
Related Publication: Yu et al. Science, 2008; Vo et al. Cell, 2016; Wierbowski et al. PNAS, 2020 -
Mapping viral-human interactomes to fight viral infections
Our lab investigates viral-host protein interactions to identify potential therapies for viral infections. We generated a comprehensive SARS-CoV-2-human protein interactome, validating known host factors and discovering novel ones. Using network-based drug screens, we identified 23 drugs with significant proximity to SARS-CoV-2 host factors, including carvedilol, which shows clinical benefits and antiviral properties.
Related Publication: Zhou et al. Nature Biotechnology, 2022 -
Constructing full-proteome multiscale 3D interactomes to identify the interfaces for all protein-protein interactions
We focus on developing machine learning frameworks for predicting partner-specific protein binding interfaces. Our previously established tool, Interactome INSIDER, has successfully provided interactome-wide protein-protein interface predictions. We are now working on PIONEER, a deep learning approach that leverages available structure information alongside sequence data, incorporating comprehensive single-protein and partner-specific features to further enhance the accuracy of protein interface predictions.
Related Publication: Meyer et al. Nature Methods, 2018; Wierbowski et al. Nature Methods, 2022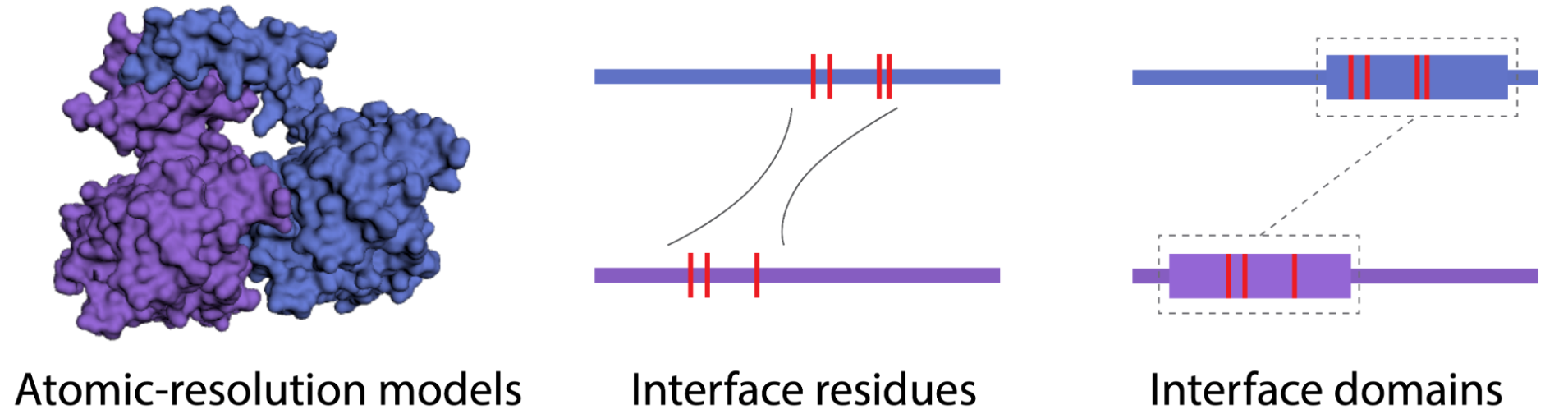
-
Understanding the impact of genomic variants on protein interactions/complexes at the whole proteome scale through computational-experimental integrated approaches
Through the combined use of computational and experimental approaches, we have analyzed thousands of missense variants and examined their effects on protein-protein interactions. Our findings reveal that disruptive SNVs are prevalent at both rare and common allele frequencies. Furthermore, our studies suggest that 10.5% of missense variants carried per individual are disruptive, a proportion higher than previously reported. This indicates that the genetic makeup of each individual may be considerably more complex than anticipated. We develop the first integrated experimental-computational interactome perturbation framework to prioritize damaging missense mutations on a genomic scale for developmental disorders, including autism.
Related Publication: Fragoza et al. Nature Communications, 2019; Chen et al. Nature Genetics, 2018 -
Functional characterization of chromatin-associated interactome networks at key regulatory steps of the transcription cycle
We aim to gain a deeper understanding of the molecular mechanisms underlying transcription regulation. Our primary objectives are threefold: (i) Investigate the transient interactions of RNA polymerase II throughout the various regulated stages of the transcription cycle; (ii) Examine the native composition and interactions of large molecular complexes involved in the regulation of transcription; (iii) Validate and functionally characterize novel factors involved in critical regulatory steps of the transcription cycle.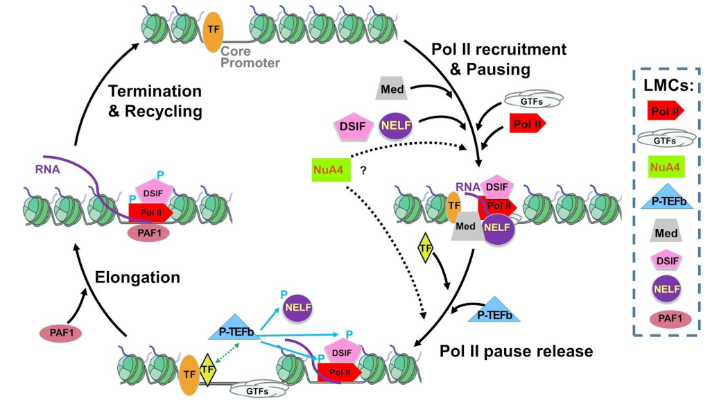
Fundamental architectures of transcriptional regulatory elements (TREs) and principles of gene regulation
-
Divergent transcription of eRNAs as a critical mark for active enhancers genome-wide
Recent studies have shown that both enhancers and promoters can recruit RNA pol II and initiate transcription. The short half-life nature of enhancer RNAs (eRNAs) makes detection of distal initiation events challenging. Through systematic comparison of RNA sequencing assays, we find that nascent transcriptome assays, PRO-cap and PRO-seq, have great sensitivity and specificity in detecting eRNA transcription genome-wide. In fact, we find that, unlike histone marks, divergent transcription of eRNAs is a critical mark for all active enhancers genome-wide. Moreover, nascent transcription precisely delineates the sequence architecture of enhancers, whereby transcription start sites (TSSs) serve as critical anchors in revealing motif positioning within enhancers and their boundaries.
Related Publication: Yao et al. Nature Biotechnology, 2022; Tippens et al. Nature Genetics, 2020
-
Generating a comprehensive landscape of transcription regulatory elements across the human body
Enhancers play a crucial role in determining cell identity by governing cell-type-specific transcriptional programs. Although the human genome contains a plethora of enhancers, only a subset of these regulatory elements are active in a particular cell type. Alterations in these regulatory elements can lead to disease phenotypes. Thus, identifying unique patterns of activated transcriptional regulatory elements, such as enhancers and promoters, can aid in understanding non-coding genetic variations associated with various diseases. Despite its importance, our understanding of active TRE usage and dynamics across the human body remains limited. Therefore, our lab aims to generate a comprehensive atlas of active TREs across the human body using near-basepair-resolution PRO-cap/seq assays. Our goal is to map and analyze the active TRE landscape in different organs, tissues, and cells of the human body at unprecedented depth and resolution. This analysis will enable us to gain a better understanding of distinct patterns and characteristics of activated enhancers and promoters and their contribution to cell-type-specific transcriptional programs. -
Understand the evolution of regulatory elements
Divergent transcription has been demonstrated as a critical mark of active enhancers. However, in addition to divergently transcribed elements, a considerable number of unidirectional elements (only transcribed in one strand) have also been detected and might also be functional in the transcription machinery. These unidirectional elements are largely overlooked and their identity and function remain elusive. We have found that unidirectional distal elements have younger sequence ages and less evolutionary constraints compared with their divergent counterparts, which suggest that they are more recently evolved and represent a distinct group of functional units. The elucidation of directionality will help build a finer architecture model and enable a better understanding of evolutionary and functional dynamics of TREs. -
Functional evaluation of key sequence features of regulatory elements and impact of noncoding variants
We are developing a range of interpretable deep learning models to capture associations between DNA sequence and run-on assays. These models are designed to distinguishing signal and sequence features of enhancers from promoter elements and designing a transcription-driven model of regulatory element identification from the genome.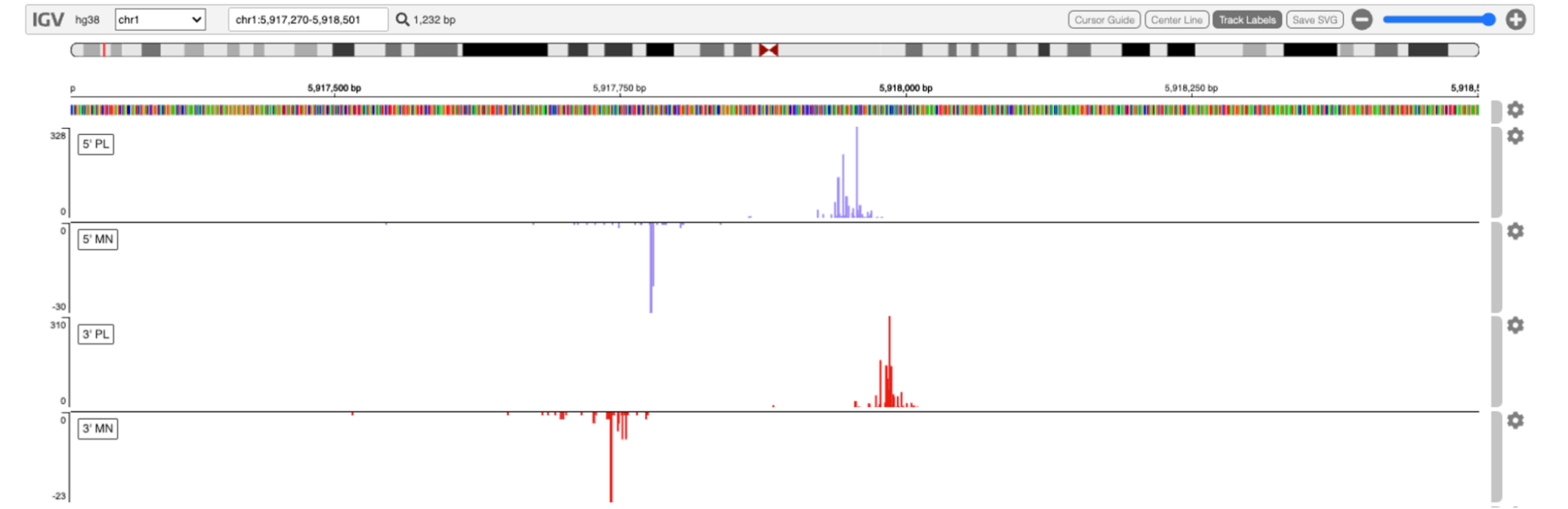
-
Enhancer-Promoter connections
Transcription initiation is a complex process that involves various regulatory mechanisms. Recent studies have demonstrated that enhancers and promoters can recruit RNA pol II and initiate transcription. While these events can serve as indicators of transcriptional regulatory elements, the underlying mechanisms of how proximal and distal initiation events interact with each other and the associated regulatory layers are not yet clear. Our lab is developing deep neural network architectures to explore the cross talks between enhancer and promoter. -
Identification of disease-associated noncoding variants
In close collaboration with Drs. Kathryn Roeder and Bernie Devlin, experts in neurodevelopmental disorders, and the Gan Lab at Weill Cornell Medicine, which employs induced Pluripotent Stem Cells (iPSCs) derived neuron cells as an in vitro model, we are capitalizing on the wealth of available genomic variant data for various human diseases, particularly autism spectrum disorder and Alzheimer's Disease. By mapping these variants to active enhancers identified by gene-distal transcription events in neuronal cell lines, we aim to construct detailed models that elucidate the functional impacts of genomic variants and their associations with phenotypic changes. This approach will help uncover the mechanisms driving disease pathogenesis, ultimately providing groundbreaking insights that advance our understanding of diseases and foster innovation in therapeutic development.
Understanding of disease mechanisms by integrating the impact of genomic variants with diverse biological networks
-
Our lab utilizes experimentally- or deep-learning-algorithm-determined protein-protein interaction interfaces, 3D protein structures, and active enhancer elements to identify potentially functional variants and mutations. Our research approach involves two key objectives: (i) investigating the molecular effects of these variants to elucidate their impact on the structure and function of proteins as well as the activity of enhancers; and (ii) expanding our understanding of their pathway-scale consequences by leveraging the wealth of information embedded within molecular networks present in cells. By combining these strategies, we aim to gain a comprehensive understanding of how genetic variations contribute to disease development and progression.